RESEARCH
ゲノムや言語をどう読み解くか?
塩基配列に隠れた情報を探し出す
─生物情報学の挑戦
さまざまな生物のゲノム配列が次々と明らかにされ報告されています。そのデータを集めたデータバンクには、毎日新しい配列が登録されています。しかし、言うまでもなく、それだけで生物がわかるわけではありません。ゲノム配列を眺めてみたところで、A、T、G、Cがただ延々と並んでいるだけで、わかるのは、せいぜいゲノムの大きさ程度です。これは、コンピュータプログラムで言えば、意味不明の0と1の配列(マシン語)しか知らされていないのと同じ状況です。現在のゲノム科学は、それとよく似た状況にあります。
これを生き物の理解につなげるには、まず遺伝子がどこにあるのかを知り、その遺伝子がどのような順番で読み出され、それが細胞の中でどのように働くかを明らかにしなくてはなりません。しかし、一体どこから手を付ければいいのか。それがハッキリしません。まだ、わからないことが多すぎるのです。そこで、いろいろな方向から研究が進められていますが、何はともあれ、ゲノム配列の中から遺伝子に当たる部分を探し出す必要があります。
見ただけでは意味不明なA、T、G、Cの並びからのGene finding(遺伝子の同定)の方法は、大きくわけると3つになります。1つは、既知の遺伝子と類似した配列を探し出しそれを遺伝子として推測するもので、非常に信頼がおける方法です。既知の遺伝子としては、実験で遺伝子としての働きが確認された他の生物のDNA塩基配列のほか、DNAからアミノ酸に翻訳すると既知のタンパク質アミノ酸配列データと一致する領域があります。2つ目は、いろいろな生物のゲノムを比較し、配列が保存されている領域を遺伝子として推測する方法です。重要な働きをしている遺伝子は、進化の過程である程度配列が保持されているという事実を利用するわけです。マウス(哺乳類)やフグ(魚類)など、ヒトゲノムと比較すると面白そうなゲノムが決定されつつあるので、今後注目される方法です。そして、3つ目が、配列がもっているパターンから遺伝子を同定する方法で、その最大の特徴は、未知の遺伝子が発見できるという点にあります。情報科学者にとって最もチャレンジングで最も関心があるのがこの方法なので、多くの人が取り組んでいますが答えはそう簡単には出てきません。
我々を含む多くの研究者が、「確率モデル」を利用してこの3つ目の方法にチャレンジしています。
ある塩基配列が端から順番にモニタに映し出されているとします。この配列がランダムであれば、A、T、G、Cは前後の配列とは無関係にいつも1/4、つまり25%の確率で出力されるはずです。しかし、Gの次はCになりやすいとか、ATTは組み合わせとして意味があるとかいうように前後の配列に依存していれば、その依存の仕方にしたがって出てくる確率は違うはずです。その規則が確率モデルなのです。たとえば、出力される確率が、直前の塩基にのみ依存している場合、定常1次マルコフモデルという確率モデルがその配列を特徴づけることになります。
そこで、遺伝子領域と非遺伝子領域、遺伝子の中でもエクソン領域とイントロン領域などそれぞれの配列に最適な確率モデルを推定する必要があります。たとえば、イントロン領域(関連記事:生命誌29号「遺伝子の中の厄介者 イントロンはどうしてなくからないか:大濱」)の配列を集め、その配列群から最適な確率モデルを推定しておけば、その確率モデルは、イントロン領域の配列を入力したときに最も高い確率で正しいA、T、G、C配列を出力することになります。逆に、ある配列が、この確率モデルによって高い確率で正しく出力された場合には、この配列はイントロン領域と考えてよいことになります。
遺伝子には3塩基を1つにしたコドンという単位があります。1コドンが1つのアミノ酸に対応するので、ここには3塩基ごとに何らかのパターンがあるはずです。3塩基単位に特有のパターンが潜んでいるのが、遺伝子がとっている一つの状態だと言えます。このようにある情報の並びの中にパターンとして潜んでいる状態を「隠れ状態」と呼びます。エクソンやイントロン、プロモーターやスプライス部位など、遺伝子の構造にかかわる単位や、塩基上の位置などは、すべて隠れ状態でゲノムの中に入っていることになります。
今度は、モニタに3塩基ずつ出力させる場合を考えます。ここに現れる3塩基が、たとえば、直前の6塩基(2コドン分)と塩基配列全体の中での位置とに依存していたとすると、この配列は、非定常2次“隠れ”マルコフモデルという確率モデルで特徴づけられます。これは、隠れ状態の移り変わりを特徴づける確率モデルです。
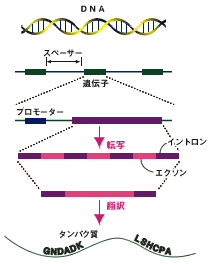
ゲノムは、非遺伝子領域、エクソン、イントロンなどさまざまな構造が組み合わさってできています。ゲノム情報科学はこれらを同定していきたいわけですが、そのためには、さまざまな構造に対応する内部状態をもつ確率モデルが必要です。内部状態としていろいろな確率モデルをもつ確率モデルがいるわけです。そしてつぎに、配列のどの部分が確率モデルのどの内部状態に対応するかを計算し、配列にラベル付けしていきます。
我々の多重出力隠れマルコフモデルをベースに使った方法で遺伝子を同定すると、原核生物のゲノムの場合には、90%以上の確率、真核生物では70%程度の的中率で遺伝子を探し出せます。今後モデルを改良し、次々と遺伝子を探し出していくのが当面の狙いです。
遺伝子はどこでしょうか?

遺伝子はここです。