2020/05/12 SRA について追記
2020/05/19 trimmomatic と fastp の結果の比較を追記
2021/03/19 初学者が迷うことが多い部分に解説を追加
シークエンスデータのQC(品質確認)・トリミング・アセンブル
シークエンスの生データには、解析対象のDNAに付加されたアダプターの配列が含まれている。また、読み取られた塩基配列(リード)には、信頼性が低い部分が含まれている。アダプター配列の存在やリードの品質を確認する作業をQC(quality check)という。バラバラに読み取られたリードを元の遺伝子配列につなぎ合わせるアセンブル作業を行う前に必ずQCを行い、アダプターや低品質な配列をデータから取り除くトリミング作業を行う必要がある。このマニュアルでは、コマンド入力をするところという意味で「:>」という記号を行頭につける。この記号は除いて、後のコマンドを改行せずに一行で入力する。
用語解説
ディレクトリ: UNIXやLinuxの場所のこと。MacやWindowsではフォルダー。ホームディレクトリ: ターミナルアプリを開いたら、一番最初にいる場所。macOSでは「/Users/アカウント名」。記号では「~/」と表記する。
パス: ディレクトリ構造によって示される場所のこと。「データのディレクトリ」と言った場合、「データが入っているフォルダーの場所」という意味。
ペアエンド: 一つのDNA断片に対し、5' 側と3' 側の両末端からシークエンスを読んだデータのこと。どちらかだけの場合は「シングルエンド」。
使用するソフトウエア
fastqc: クオリティチェックtrimmomatic: アダプターと低品質配列を除去する
fastp: fastqc + trimmomatic の機能を一本で持っていて、しかも高速
Trinity: RNA-seq データのアセンブルを行う
0 シークエンスリードのクオリティチェック
FastQファイルを解析用HDDにコピーする(MiSeqの出力データの例)
/HDD/シークエンスフォルダー/Data/Intensities/BaseCalls/*.fastq.gz
「*」は適宜自分が設定したファイル名に読み替える。
SRA (Sequence Read Archive) からデータを取得して解析に利用する場合、ファイルのヘッダー表記の問題で Trinity 実行中にエラーが起きる。Trinity の公式FAQに、NCBI の SRA Toolkit を使った解決方法が紹介されているので、以下の手順に従って FASTQ ファイルに変換する。
準備
NCBI SRA Toolkit を Anaconda を使ってインストールする
:> conda install sra-tools
SRA Toolkit に含まれるツールを使って SRA ファイルを取得し、FASTQ に変換する
:> fasterq-dump --threads 8 --defline-seq '@$sn[_$rn]/$ri' --split-files file.sra
上記コマンドの file.sra の所を、取得したい SRA ファイルの名前(SRR00000等)に変えて実行する
threads の数は、実際に使っているMacのCPU数に合わせる
fasterq-dump は fastq-dump の後継ソフトで、マルチスレッド化されているため高速に処理が可能
SRA ファイルは FASTQ への変換後も削除されないので、ストレージを圧迫する場合は手動で削除する
解析用フォルダーに移動する
ここでは、自分のホームディレクトリ(/Users/アカウント名 または ~/ と表記。「~[ちるだ]はホームディレクトリを意味する記号)に作った「Analysis」というフォルダーの中にある「fastq」というフォルダーにシークエンスデータを入れたものとして説明する。
:> cd ~/Analysis
「cd」はchange directoryの略で、ディレクトリ(場所)を移動するときに使うコマンド。
以下、コマンドは「ディレクトリ(今いる場所)」を基準にしてファイル等に操作を行うことになるので、ディレクトリはいつも意識する必要がある。コマンドライン操作に不慣れな人は、ディレクトリを意識するというところがうまくできずに迷子になってつまずくことが多い。
QC結果出力用フォルダーを作る
:> mkdir QC_out
mkdir は新しいディレクトリ(フォルダー)を作るコマンド
fastqcを使ってクオリティチェックする場合
:> fastqc fastqファイル -o 結果出力フォルダー
例: :> fastqc ./fastq/test_R1.fastq.gz -o ./QC_out
fastqファイルがたくさんある場合は面倒なので、シェルコマンドで自動化する
:> for file in `ls ./fastq/*`;do fastqc -o ./QC_out $file;done
for i in j;do コマンド;done
という命令で、「do~;done」で挟まれたコマンドをjで指定した回数繰り返すという意味になる。上記の例では、forで繰り返しを指示して、「file」という名前の箱を用意、inで「ls(リストを表示するコマンド)」の実行結果(fastqフォルダに入っているファイル)についてdoで指定したコマンドを自動で繰り返す。doの中ではfastqcを動かして、「$file」でfor文で用意した「file」という箱の中身を対象ファイル名として与えている。「file」には「ls」で表示したファイルの名前が入っている。
for文で繰り返し処理を行う際、バッククォート「`」で囲んだコマンドの結果を代入して利用できる。バッククォート「`」は、Apple製 JISキーボードでは shift + @ なので、シングルクォート「'」(shift + 7)と間違えないように気をつける。以後、このマニュアル内でも何度も登場するし、for文による自動化は様々な場面で便利なので、覚えておこう。
QC_out にhtmlファイルができているので、ウェブブラウザーを使って fastq ファイルの品質を確認できる。
fastpを使ってクオリティチェックする場合
:> fastp -i fastqファイル_R1 -I fastqファイル_R2 -h 結果出力フォルダー/report.html -j 結果出力フォルダー/report.json
例: :> fastp -i ./fastq/test_R1.fastq.gz -I ./fastq/test_R2.fastq.gz -h ./QC_out/report.html -j ./QC_out/report.json
fastqファイルがたくさんある場合は面倒なので、シェルコマンドで自動化する
:> for file in `ls ./fastq/*`;do fastp -i $file -h ./QC_out/$file_report.html -j ./QC_out/$file_report.json;done
詳細は下記1bを参照
QC_outフォルダーの中に出力されたHTMLファイルをブラウザーで開いて確認
1a 低品質シークエンスのトリミング trimmomatic
アダプター配列のファイルにリンクを作成する
:> ln -s /Analysis/Trinity-v2.10.0/trinity-plugins/Trimmomatic-0.36/adapters/TruSeq3-PE-2.fa ./
「ln」はリンク(エイリアス)を作成するコマンド。trimmomaticは、除去するアダプターの配列を書き込んだファイルを指定する必要があるが、実行するディレクトリーにないと読み込んでくれないので、今いる場所にリンクを作成する。
出力結果を保存するフォルダーを作る
:> mkdir trimmed trimmed_QC trimmed_summary
mkdir コマンドは、スペースで区切ると複数のディレクトリーを同時に作成できる
使い方
trimmomatic PE -threads 数字 -phred33 -summary ./保存場所/ファイル名 ./保存場所/リード1.fastq.gz ./保存場所/リード2.fastq.gz ./出力保存場所/ペアエンドで残ったリード1_P.fastq.gz ./出力保存場所/ペアエンドの相手が残らなかったリード1_U.fastq.gz ./出力保存場所/ペアエンドで残ったリード2_P.fastq.gz ./出力保存場所/ペアエンドの相手が残らなかったリード2_U.fastq.gz ILLUMINACLIP:TruSeq3-PE-2.fa:2:40:15 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:20 MINLEN:50
ILLUMINACLIP: アダプター配列のファイルと、トリミングの各種パラメーターの設定。
全てのリードファイルを自動で処理する
:> ls ./fastq/*R1*|sed -e ’s/fastq\/\(.*\)_R1.*/\1/g' > FastQ_list.txt
例えば、「sample_L001_R1.fastq.gz」「sample_L001_R2.fastq.gz」という名前のリードファイルがあった場合、R1やR2から後ろが削除された「sample_L001」とだけFastQ_list.txt内に書かれている。
sedコマンドの検索上分を挟んでいる点は、シングルクォートです。Apple製JISキーボードではShift+7。この画面からコマンドをコピペした場合はエラーになることが多いので、シングルクォートは手動で入力し直した方が良いです。
:> for file in `cat FastQ_list.txt`;do trimmomatic PE -threads 16 -phred33 -summary ./trimmed_summary/summary_${file}.txt ./fastq/${file}_R1_001.fastq.gz ./fasatq/${file}_R2_001.fastq.gz ./trimmed/${file}_R1_P.fastq.gz ./trimmed/${file}_R1_U.fastq.gz ./trimmed/${file}_R2_P.fastq.gz ./trimmed/${file}_R2_U.fastq.gz ILLUMINACLIP:TruSeq3-PE-2.fa:2:40:15 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:20 MINLEN:50;done
「./fastq/${file}_R1_001.fastq.gz」の部分は、cat コマンドで読み込んだFastQ_list.txtの内容の各行の前後に「./fastq/」と「_R1_001.fastq.gz」を追加して「./fastq/sample_L001_R1_001.fastq.gz」というファイルを読み込む。同様に「./fasatq/${file}_R2_001.fastq.gz」では「./fastq/sample_L001_R2_001.fastq.gz」というファイルを読み込む。
catコマンド前後の点は、バッククォートです。Apple製JISキーボードでは Shift+@。この画面からコマンドをコピペした場合はエラーになることが多いので、バッククォートは手動で入力し直した方が良いです。
よくある間違いは、上記コマンドの「./fastq」の部分を自分がファイルを置いたディレクトリーの名前に変更していないため、file not found というエラーが出てしまうことである。
トリミングされたファイルを FastQC で確認する。方法は上の内容と同じ。出力先をQC_outからtrimmed_QCに変更する。
・trimmomatic は Trinity のプラグインとして標準搭載されているトリミングソフト。
・上記のコマンドでは、trimmed に綺麗になったシークエンスファイル、trimmed_summary にトリミングの結果何%のシークエンスデータが残り、何%のデータが除去されたか確認できる。
・-threads では、自分が使っているMacのCPU数を指定する。
・ILLUMINACLIP では、アダプターの配列ファイルを指定する。HiSeqやMiSeqを使った場合はTruSeq3-PE-2.faを指定する。
1b 低品質シークエンスのトリミング fastp
fastp は、上記の fastqc と trimmomatic の機能を併せ持ったソフトウエアである。しかも、上記二つと比較して、動作速度が圧倒的に速いという特徴があるのでオススメである。fastp と trimmomatic + fastqc では、後者の方が実績があるものの前者の方が便利なので、これから使い方を学ぶ初学者は、fastpを使うと良いのではないだろうか。
出力結果を保存するフォルダーを作る
:> mkdir trimmed trimmed_QC
mkdir コマンドは、スペースで区切ると複数のディレクトリーを同時に作成できる
使い方
fastp --detect_adapter_for_pe -i ./保存場所/リード1.fastq.gz -I ./保存場所/リード2.fastq.gz -o ./出力保存場所/リード1_P.fastq.gz -O ./出力保存場所/リード2_P.fastq.gz -h ./QCファイル保存場所/report.html -j ./QCファイル保存場所/report.json -w 数字
--detect_adapter_for_pe: fastpはアダプターを自動認識して除去する機能を持っているが、ペアエンドの場合はこのオプションをつけないと無効になる。アダプターを除去しないとアセンブルの結果が大幅に悪化するので必須のオプション。
-i 解析したいファイルのR1
-I 解析したいファイルのR2
-o R1のトリミング結果出力ファイル
-O R2のトリミング結果出力ファイル
-h html のレポート出力先
-j json のレポート出力先
-w 使用するCPUの数
:> ls ./fastq/*R1*|sed -e ’s/fastq\/\(.*\)_R1.*/\1/g' > FastQ_list.txt
:> for file in `cat FastQ_list.txt`;do fastp --detect_adapter_for_pe -i ./fastq/${file}_R1_001.fastq.gz -I ./fasatq/${file}_R2_001.fastq.gz -o ./trimmed/${file}_R1_P.fastq.gz -O ./trimmed/${file}_R2_P.fastq.gz -h ./trimmed_QC/${file}_report.html -j ./trimmed_QC/${file}_report.json -w 16;done
trimmed_QCに造られたレポートファイルで、シークエンスのクオリティとトリミングの結果を確認する。htmlファイルは、ウェブブラウザーで開くことができる。
fastqc と同様に、クオリティチェックのみ行ってトリミングをしない場合は、出力のオプション(-o, -O) をつけずに実行する。
例: fastp -i sample_R1.fastq.gz -I sample_R2.fastq.gz
2 全てのリードをまとめてアセンブルする
テキストファイルを作る:> touch Trinity-V2100.sh
$TRINITY_HOME/Trinity --seqType fq --max_memory 80G --CPU 16 --KMER_SIZE 32 --output trinity_2100_out --left seq_read1_R1.fastq.gz,seq_read2_R1.fastq.gz,seq_read3_R1.fastq.gz --right seq_read1_R2.fastq.gz,seq_read2_R2.fastq.gz,seq_read3_R1.fastq.gz && $TRINITY_HOME/util/TrinityStats.pl ./trinity_2100_out/Trinity.fasta > ./stats_2100.txt
--seqType: シークエンスデータのファイルタイプを指定。普通はFASTQだと思うので、fq を指定。
--max_memory: Trinity が使う実メモリーの最大値。Macに搭載しているメモリから、システムが使用する分を引いて少し余裕を持たせるくらいが良い。
--CPU: 計算に使うCPUの数。Macが搭載しているCPU数を設定。
--KMER_SIZE: アセンブル結果に大きな影響があるパラメーターであるが、バグが見つかり Trinity Ver. 2.9.1 以降では非搭載なので、このオプションは削除して使用する。早期の復活を願う機能である。
--output: 結果を保存するフォルダー名。
--left: ベアエンドサンプルのリード1。サンプルが複数ある場合は、「,」スペース無しのコンマでつなぐ。例: --left sample1_R1.fastq.gz,sample2_R1.fastq.gz,sample3_R1.fastq.gz
--right:ベアエンドサンプルのリード2。サンプルが複数ある場合は、「,」スペース無しのコンマでつなぐ。例: --right sample1_R2.fastq.gz,sample2_R2.fastq.gz,sample3_R2.fastq.gz
上記のコマンドでは、アセンブル終了後に TrinityStats.pl を使って遺伝子数やN50を計算している。
Trinity にはこの他にもたくさんのオプションがあって、徹底的に追い込むとかなり凝った設定も可能になる。全てのオプションをみたい場合は
$TRINITY_HOME/Trinity --show_full_usage_info
とする。
FastQファイルがたくさんある場合
:> ls ./fastqフォルダー/*R1_P* | gsed -z 's/\n/,/g' > left.txt
:> ls ./fastqフォルダー/*R2_P* | gsed -z 's/\n/,/g' > right.txt
ls コマンドでファイルのリストを取得、パイプで gsed を使って改行をコンマに変換、テキストファイルに保存
作成したテキストファイルを開いて、Trinity コマンドの --left, --right のところに貼り付ける
:> /bin/sh ./Trinity-v2100.sh
使用するMacの性能やデータの量にもよるが、数日から数週間ほど計算にかかる。
計算が終わったら、stats_2100.txt というファイルができている。テキストエディターで開いて確認する。
目安としては、genes や transcripts の数が小さいほど良く、N50の数値が大きいほど良い。結果は生物種にもよるが、感覚的にN50が1000塩基未満であれば、あまりアセンブルの精度が良いとは言えず、遺伝子探索にも支障が出る可能性もある。アセンブルに利用するリードを増やすなど、改善の戦略に役立てる。
スクリプトファイル(.sh という拡張子をつけたファイル)を作成する意味
スクリプトファイルを作成して作業したフォルダー内に残しておくことで、作業ログにもなる。そのフォルダー内に残された解析結果が、どのようなコマンドによって得られたものなのか確実な記録になるので、完璧な再現性を保証する研究ノート代わりにもなる。紙のノートに手書きする際に発生する転記ミスを防ぐ意味もあるので、解析研究での研究ノートの残し方としてお薦めする。スクリプトは「#」から改行コードまでをメモとして扱い、コマンドには影響しないという仕組みがあるので、上記のスクリプ内に実行した日付や研究の条件などを書き込んでおくと、研究ノートとしての情報量が上がってより有益である。
例
# 2020/05/05
# ナミアゲハのRNA-seq
trimmomatic と fastp の比較
アダプターの削除と低品質配列の除去ができる両ソフトですが、どちらを使ったら良いのか迷う場合は下記を参考にしてください。同じデータを使って、 trimmomatic で前処理を行った場合と fastp で前処理を行った場合を比較。それ以外、Trinity のバージョンや設定は同じ。
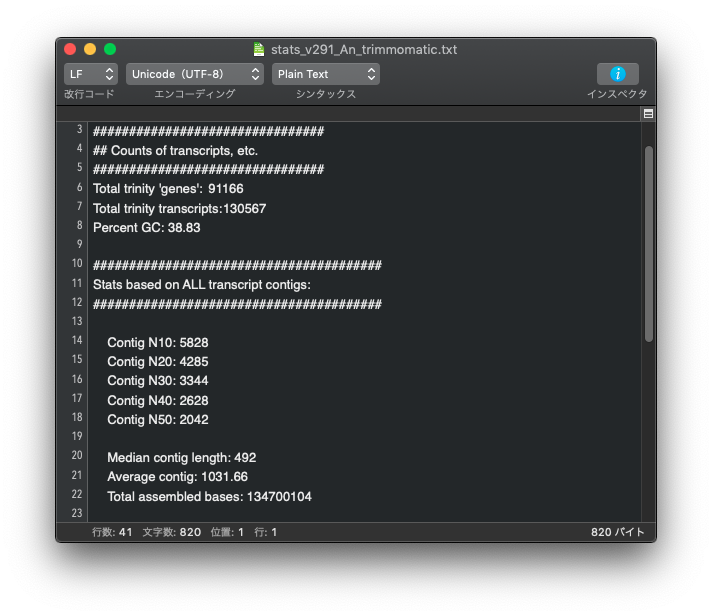
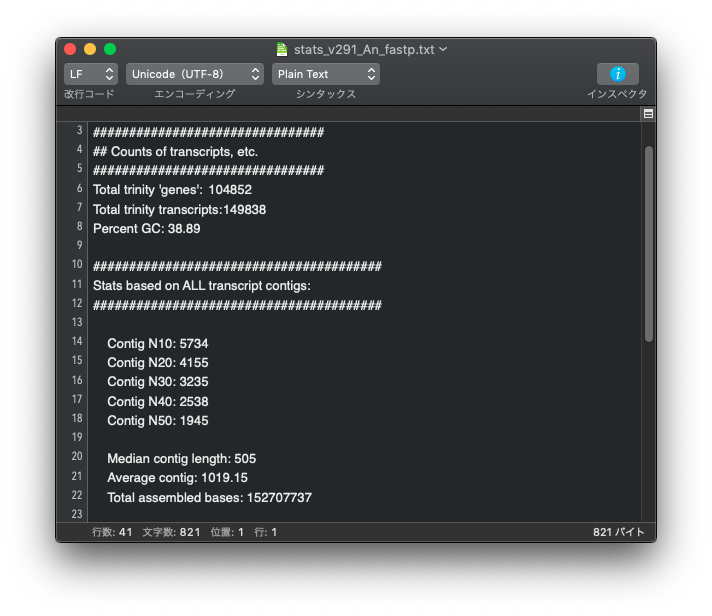
上: trimmomatic, 下: fastp
genes の数も transctipts の数も trimmomatic を使って前処理を行った方が少なくなっている。
Contig の Nx の値も、trimmomatic を使った方が長くなっている。
この結果だけを見ると trimmomatic を使った方が良好な結果が得られている。この程度の差は大したことはないと見るか、無視できない差であると考えるかは各自で判断すること。
全てのデータで同じ傾向になるかは不明であるが、一つの目安になるのではないだろうか。
目次
- 環境構築
- リードのクオリティチェックとアセンブル
- Transdecoder による遺伝子予測とアノテーション
- リード数のカウント
- カウントマトリクスの作成
- リードカウントのQC解析
- DE解析
- DEG取り出し
- おまけ1: Transdecoderの自動化
- おまけ2: 発現量解析の自動化
- おまけ3: Anacondaコマンドの使い方一覧